生命動態のデータサイエンス(DS2)
(春学期 火曜日3時限) 慶應義塾大学 政策・メディア研究科 後期博士課程 教育体験 講義資料 (Last updated on June 1st by Daniel Evans-Yamamoto)
内容:COVID19の原因ウイルスSARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2)について、公開データをもとにゲノムデータをダウンロードし、タンパク質配列への翻訳および疫学データから得られる変異情報と対応させる((第1回) 。第2回では、タンパク質の立体構造データをProtein Data Bank (PDB)から取得し、立体構造上で変異情報を描画する。最後に、第3回では、ウイルスのタンパク質とホストのタンパク質の結合面を確認し、変異がそのようなドメインにあるのかや、タンパク質間相互作用ネットワークについて確認する。
評価は、各回に課される課題に基づいて行う。
教育体験 第1回 2020/6/2
目次
- Introduction
- SARS-CoV-2のゲノムをダウンロード
- ORF領域を抽出、翻訳
- SARS-CoV-2株間の変異情報の確認
- SARS-CoV-2の変異情報を確認しよう(演習課題)
0 自己紹介

1.1 Introduction
本講義では、現在世界中で猛威を奮っているSARS-CoV-2ウイルスを題材に、「変異」とは何か、また機能にどのように影響を及ぼすのかについてその解析の一例を体験する。この過程で、DNAに起きた変異がどのように形態の違いに寄与するかなどを理解し、今後の研究等に役立てていただきたい。
まず、SARS-CoV-2ウイルスを見てみよう。
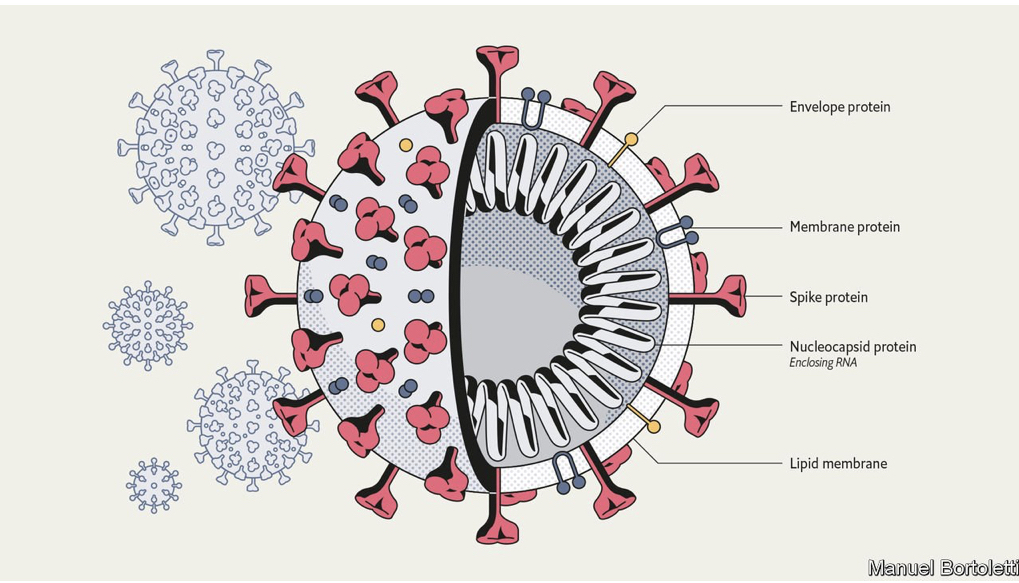
このウイルスがどのようにヒトに感染するかを知る前に、セントラルドグマについてざっとおさらいしよう。セントラルドグマとは、DNAからRNAが転写され、そのRNAを翻訳してタンパク質が作られるという生物におけるルールだ。このタンパク質は遺伝子によって多種多様にあり、細胞の機能・維持に主として働いていることも付け加えたい。

このセントラルドグマをハックするのがウイルスや、バクテリオファージ(細菌に感染するウイルス)だ。ウイルスは、単体では自己複製することができず、宿主の転写・翻訳機構を使い自己を複製する。
余談として、広く浸透している生物の定義は以下の3つを満たすものである。
- 外界と膜で仕切られている。
- 代謝(物質やエネルギーの流れ)を行う。
- 自分の複製を作る。
ウイルスは1と2を満たしているが、3 はどうだろう? 宿主を利用して自己を複製するが、単体ではできない点で生物に含めるか否かの議論が依然として行われている。
次にSARS-CoV-2がどのように宿主に感染するかを見てみよう。このウイルスのゲノムは14のタンパク質をコードしており、それぞれ機能を担っている。
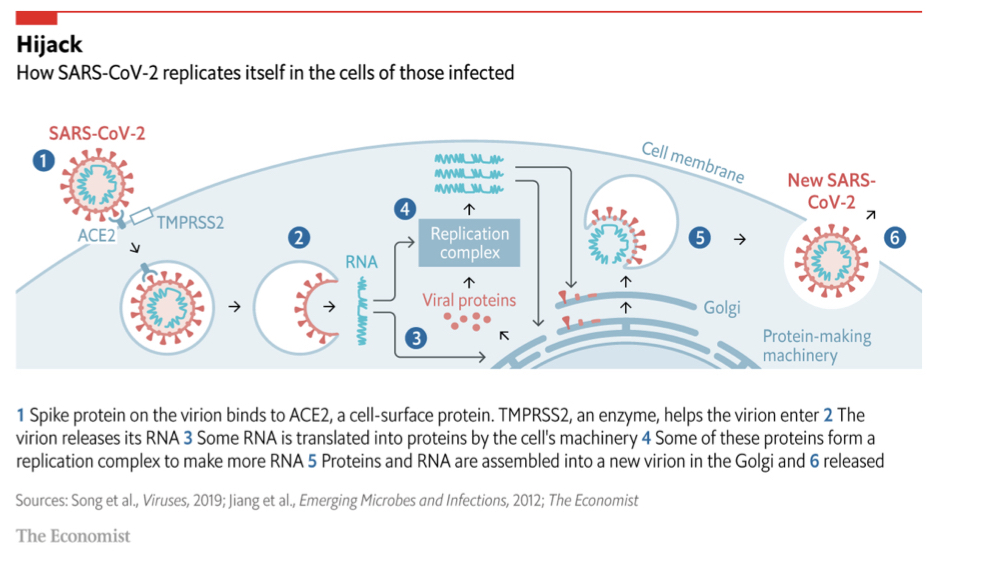
1.2. SARS-CoV-2のゲノムをダウンロード
NCBIからDNA配列を取得する。SARS-CoV-2のNCBI accessionは武漢で単離されたNC_045512で、これはRefSeqと言われるレファレンスに指定されている。NCBIのSARS-CoV-2ページにはこれに加えて世界中で採取された5,000近い配列が登録されている。
ここでは先週行った、Rのseqinrパッケージを用いたゲノムのダウンロードを行う。
library(seqinr)
ACCESSION <- "NC_045512" # SARS-CoV-2
filename <- paste0("https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=",ACCESSION,"&rettype=fasta&retmode=text")
seqs <- read.fasta(file=filename, seqtype="DNA", strip.desc=TRUE)
sarscov2 <- seqs[[1]]
1.3. ORF領域を抽出、翻訳
SARS-CoV-2ゲノム中には、14の機能的タンパク質がコードされていることが推測されている。今回はSARS-CoV-2の中でも、ヒト細胞に感染する際に重要なSpike proteinに着目する。 タンパク質がコードされている配列位置情報はNCBIのWEBサイト(https://www.ncbi.nlm.nih.gov/nuccore/NC_045512)で確認できる。
#SARS-CoV-2のSpike Protein はゲノムの21563bpから25384bpの位置にコードされているため、これを抽出する
spike_cds = sarscov2[21563:25384]
DNAは生体内でRNAに翻訳され、その後遺伝暗号に従いアミノ酸配列に翻訳される。その対応表を下に示す。
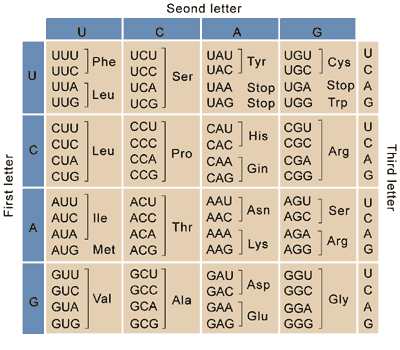
ここでは、遺伝暗号表(コドンテーブル)をもとにseqinr内のgetTrans関数で核酸配列をアミノ酸(タンパク質)配列に翻訳する。
spike_AA = getTrans(spike_cds)
#プリントしてみよう 1文字目は必ずメチオニンを表すMとなるはず
spike_AA
1.4. SARS-CoV-2株間の変異情報の確認
公開されたCOVID-19のゲノム配列にもとづく疫学データを見てみると、ウイルス株の変異パターンから流行の過程を推測することができる。そのような変異はゲノム上のどのような位置にあるのだろうか?
これを踏まえて、「変異している=より感染力が強い」と結論付けられるのだろうか?
進化とは、この多様性の獲得(変異;variation)と選択 (淘汰;selection)が行われることで、次第に環境に適応した個体が残ることである。選択のステップで獲得された変異が利益(あるいは)不利益を被る場合はそのような進化は助長されるが、中にはあってもなくても変わらない(中立的)変異も存在する。したがって、変異があるからと言ってそれがすなわちより危ないウイルスであるということでは決してない。
SARS-CoV-2ではテクニカルなアーティファクトを含む多くの変異が報告されているが、本講義ではそれらに着目し、機能変化を実現し得るかを確認してみよう。
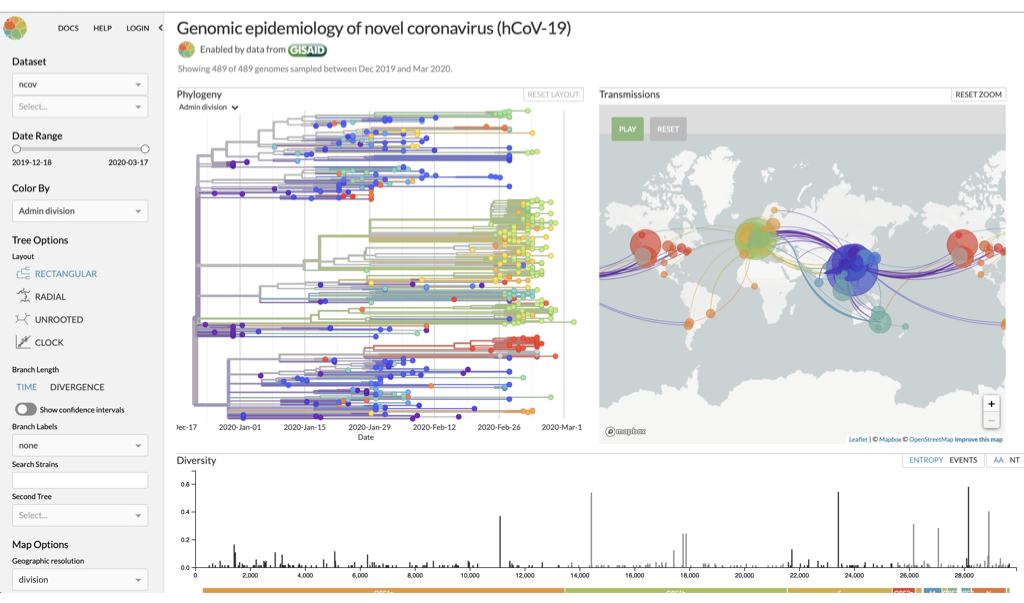 この二つの株間では機能に違いはあるのだろうか?
この二つの株間では機能に違いはあるのだろうか?
1.5. SARS-CoV-2の変異情報を確認しよう(演習課題)
疫学データをもとに、ゲノム上の変異位置の一つに着目しよう。
そこで、
- 選んだ変異がどこでサンプリングされたか
- どのタンパク質コード領域か
- タンパク質をコードする何番目のアミノ酸か を確認しよう。
確認されたサンプリングポイントのゲノムをNCBIのSARS-CoV-2ページで見つけ、IDを控えよう。
1.2 ~ 1.3と同様に下記手順に従って着目した株の変異情報を見てみよう
MUTATION <- "MT320538" # SARS-CoV-2, France
filename <- paste0("https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=",MUTATION,"&rettype=fasta&retmode=text")
seqs <- read.fasta(file=filename, seqtype="DNA", strip.desc=TRUE)
sarscov2_mut <- seqs[[1]]
#ここではSpike protein に着目しゲノムの21563bpから25384bpを抽出する
spikemut_cds = sarscov2_mut[21563:25384]
#翻訳
spike_AA_mut = getTrans(spikemut_cds)
実際にどんな変異が入っているか比較してみよう。着目したい「pos」は疫学データをもとに入れてみよう。アミノ酸配列でのposはタンパク質をコードする何番目のアミノ酸かをもとにしよう。念のため幅を持って[pos-15:pos+15]のように表示しよう。
#核酸の変異はあるか?
spike_cds[pos]
spikemut_cds[pos]
#アミノ酸配列の変異はあるか?
spike_AA[pos]
spike_AA_mut[pos]
ここまでの出力内容を課題として提出しよう。 期限は2020/6/8 23:59
効率的にに配列間の違いを確認する方法として、しばしば配列間アラインメントを行います。この内容は次回の鈴木さんの講義でカバーされる予定です。
##
##
##
教育体験 第2回 2020/6/9
タンパク質配列における変異はどのような意味を持つのだろう?
目次
- マルチプルアラインメント
- タンパク質の構造をPDBで確認
各演習の締め切りは 2020/6/15 23:59 です。
どの変異が機能に影響するのか知る術はあるのだろうか?
一般的には変異はランダムに導入される。例えば、あるタンパク質についてその変異体全ての機能を測定することはできるだろうか?
タンパク質の機能について少し考えてみよう。
タンパク質には、以下のようなものがある。
- 酵素
- 複合体タンパク質
- DNA/RNA結合タンパク質
酵素
タンパク質には、酵素と言われる代謝物や有機物を触媒するものがある。このような酵素は有機物との接地面にその酵素たらしめる機能が備わっていることになる。ではそのような位置に変異を導入すれば酵素活性を失活させることができるということになる。逆に、自然界でその酵素が生育に重要な役割を果たしていると、そのような部位に変異が入った個体は死んでしまい、我々には観測でにないだろう。
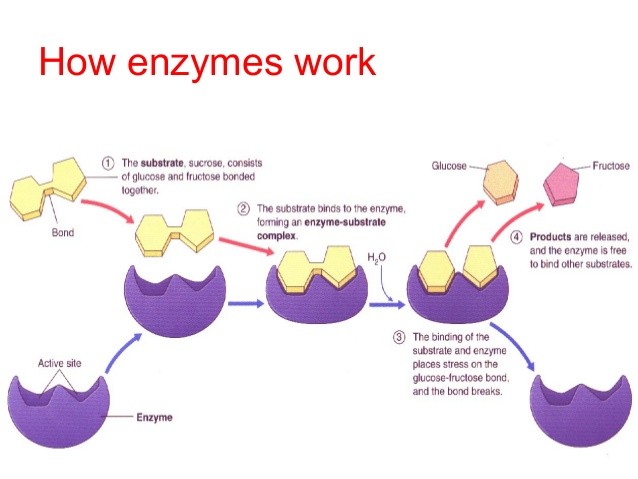
複合体タンパク質
タンパク質は生体のほとんどの機能を担う。タンパク質をコードする遺伝子がヒトで20,000を超えることからも、その多様性を想像いただけるだろうか。その多様性を持ってして、それぞれ別々の機能を担っているが一つのタンパク質が単体で機能を担うことはあまりない。それよりも、複合体を形成したり他のタンパク質と協調して機能を達成する。このような場合、相互作用面に変異が入ったらどうなるだろうか?鍵と鍵穴の関係を考えてもらうと、わかりやすい。家の鍵をお隣さんと交換したら自分の家(元の相互作用相手)は開かない。でも、お隣さんの家(違う相互作用相手)は開けることができる。ただ全く違う家に対してはそうもいかないため、鍵の特異性は保ったままである。(わかりにくいかな、、、、)要するに言いたいことは、お互いが相互作用を維持するように少しだけ変わることで変異を蓄積していくことができ、実際自然界ではそのように観測される。この現象は共進化(Coevolution)と言われる。この変異の獲得のスピードはタンパク質がどれくらい変異に頑健か等の要因によって決まるため、ほとんど配列の変わらないものもあればとても変容するものまで様々だ。

DNA/RNA結合タンパク質
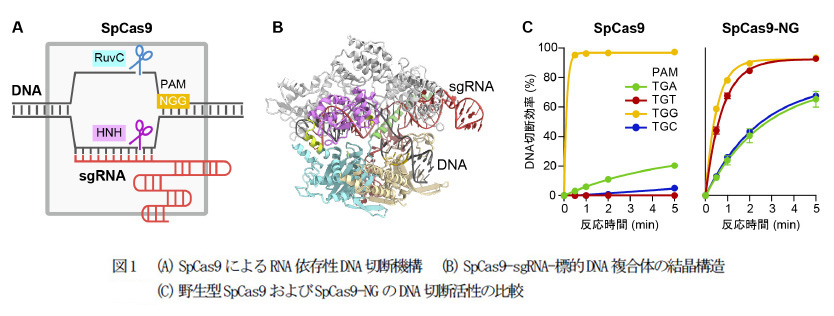
タンパク質の中には、RNAやDNAに結合して切断や修飾を行うものもある。有名なものにCRISPR/Cas9でお馴染みのCas9タンパク質がある。S. pyogenes由来のこの機構は従来は外来DNA(ウイルス/ファージ)に対する免疫システムとしてあり、近年ではゲノム編集等に広く使われている。このタンパク質はsgRNAというRNAに結合し、そのRNA配列とマッチするDNAを切断する。ただ、Cas9がDNAに結合するドメインが、PAMと言われる制約を持つ(spCas9の場合はGGなので、理論的には16塩基に一回編集できる場所がある)。東京大学の西増准教授らは、このPAMの制約を減らすような7つの変異を(それまでの経験と類稀なる選眼で)選定し、この制約をGのみ(理論的には4塩基に一ヶ所)に減らすことに成功した。ただ、そのようなプロ技で全てのタンパク質を最適化することは不可能に近い。
Deep Mutational Scanning
タンパク質の機能を測定する上で、合成生物学的なアプローチを用いて人工的に変異を導入し、細胞成育から重要な残基を特定する手法がある。これはDeep Mutational Scanningと言われ、多くのバリアント(例えば300アミノ酸長であれば、300-1 x 20 = 5980通り)を一斉にスクリーニングするため特定の一つのタンパク質の特定の機能に注目して解析される。

上記を踏まえて、この講義では自然界にある変異がどのような箇所に入っているのか、また機能ドメインと関連性があるのかについて(1)マルチプルアラインメントと(2)立体構造をもとに考察しよう。
1. マルチプルアラインメント
比較解析を行う上で、配列や構造のアラインメントが行われる。アラインメントには大きく分けて「ローカルアラインメント」と「グローバルアラインメント」があり、前者はデータベースから何が相同な配列であるかや部分的なマッチを探すのに適しているのに対して、後者はすでに一程度相同な配列に対して全長の違いを見るのに用いられる。ここではこれを複数の配列を用いるマルチプルアラインメントを行ってみよう。
アミノ酸のマッチはその極性等の性質の近さから下記の表によって得点化される。
マルチプルアラインメントのパッケージ”msa”に沿って進めてみよう。
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("msa")
library(msa)
テストデータをロードし中身を確認しよう。
mySequenceFile <- system.file("examples", "exampleAA.fasta", package="msa")
mySequences <- readAAStringSet(mySequenceFile)
mySequences
アラインメントを行う。
myFirstAlignment <- msa(mySequences)
#use default substitution matrix
#結果の表示
myFirstAlignment
#Plotのtexファイルを出力
msaPrettyPrint(myFirstAlignment, output="pdf", showNames="none", showLogo="none", askForOverwrite=FALSE, verbose=FALSE)
ここで出力される”myFirstAlignment.tex”をみるにはtexとLatexを用いる必要があるので、適宜ダウンロードしよう。
!!注意!!
Tex/LaTexは数Gbあります。通信環境に余裕がない場合は、Slackにてファイルを送ってください。こちらで描画して図を送ります。

演習:マルチプルアラインメント
テストデータの出力が確認されたら、自分のデータで行う演習に入ります。
#以下のようなmulti fasta ファイルを用意しよう。
>NC_045512
MFVFLKGVKLHYTMFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFR
>MT578015
SSVLHSTCSCLKGCCSCGSCCKFDEDDSEPVLKGVKLHYT
>MT539729
MFVFLVLLGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDDSEPVLKGVKLHYT
ここでSlackに自分の対象(題材)を送ってみよう。 どこで配列を取得するか・Fastaにする方法をガイドするので、自分の題材にあった配列を取得しよう。
#データをロードする
mySequences <- readAAStringSet(file.choose())
#ポップアップが出るので、自分のファイルを選択しよう。
#配列を確認する
mySequences
#アラインメントを行う。
myAlignment <- msa(mySequences)
#use default substitution matrix
#結果の表示
myAlignment
#Plotのtexファイルを出力
msaPrettyPrint(myAlignment, output="pdf", showNames="none", showLogo="none", askForOverwrite=FALSE, verbose=FALSE)
このファイルをTex/LaTexで開くと、マルチプルアラインメントの図が見られます。
2. タンパク質の構造をPDB上で確認しよう
ここではまず初めにSARS-CoV2のSpike proteinを例にProtein Data Bank で公開されている立体構造を確認し、マルチプルアラインメントの結果との比較を行います。その後、それぞれの題材に近い立体構造について同様のことを演習として行います。
Protein Data Bankにアクセスしよう。
演習:PDB 立体構造
Protein Data Bankにアクセスし、好きなタンパク質を調べよう。
そのタンパク質について、3D veiwで表示しよう。
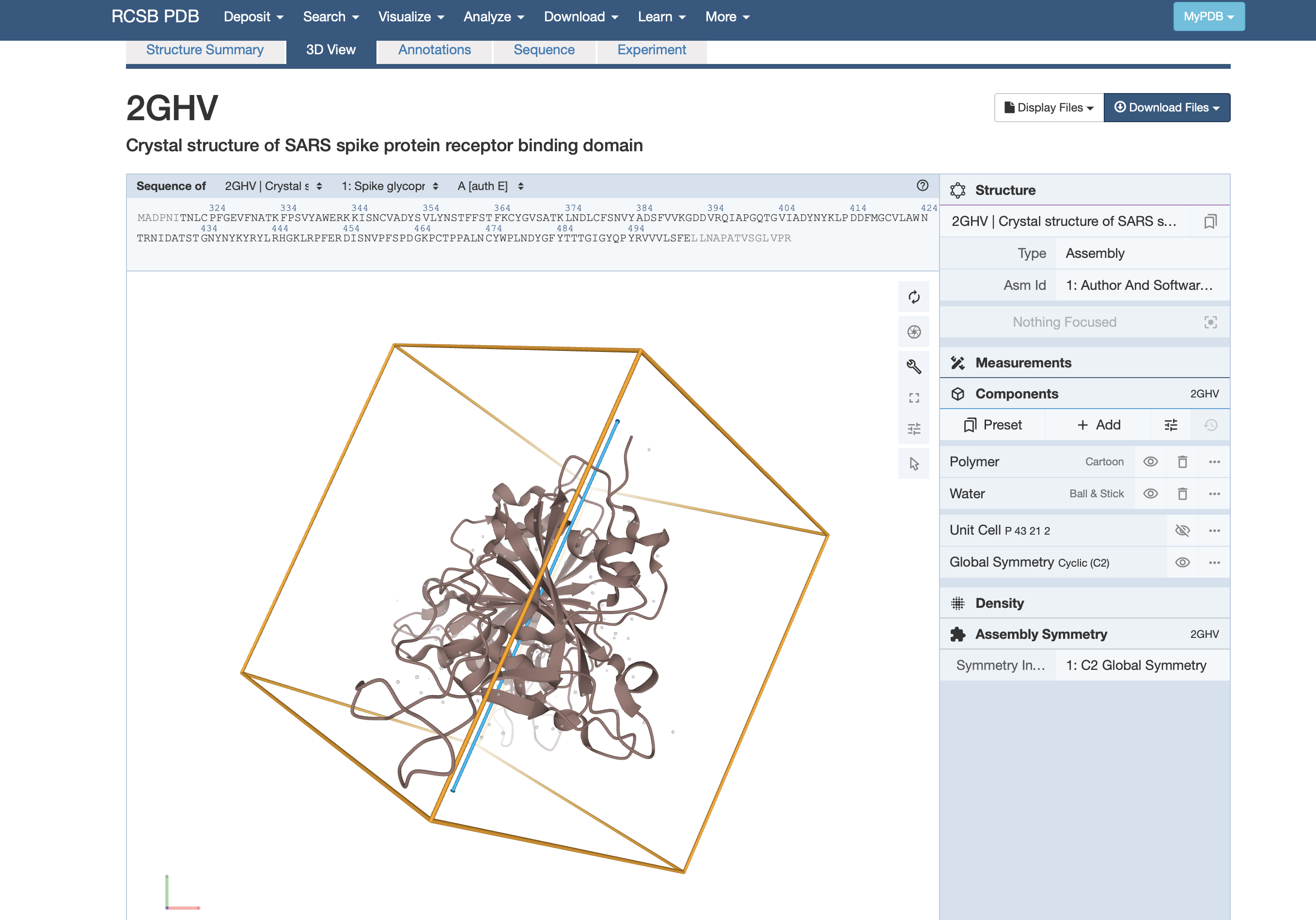
- マルチプルアラインメントで着目した位置をハイライトしてみよう。
- 極性のあるアミノ酸をハイライトしてみよう。
## ##
教育体験 第3回 2020/6/15
タンパク質間相互作用を可視化しよう
目次
- タンパク質間相互作用データベースにアクセスしよう
- 着目するタンパク質について、その相互作用を確認しよう
- 第2回で確認したバリアントの位置を照らしてみよう (演習課題)
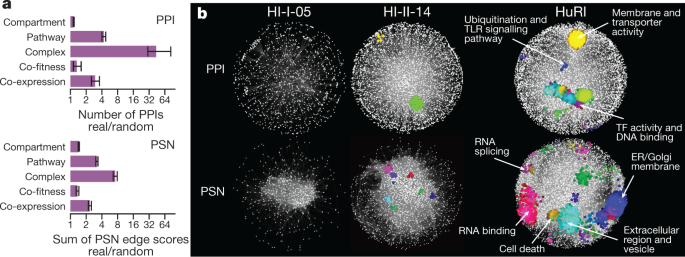
第2回で紹介したように、タンパク質は相互に作用して細胞の機能を担い、細胞の複雑な表現型を実現する。2020年に発表されたヒトのレファレンスインタラクトーム(HuRI,下図)では、12,000のタンパク質の全対全ペアの相互作用をマップを構築している。ここでは出が酵母を用いた手法であるY2H法が用いられた。
このようなタンパク質相互作用データはデータベースに蓄積されており、モデル生物をはじめとする多くが登録されている。第3回では、これまでに各々が着目したタンパク質(群)についてタンパク質間相互作用データベースにアクセスし、その相互作用を確認するとともにそのうちのいくつかについて立体構造を確認する。
そもそも「相互作用」とはなんだろう?タンパク質や核酸の分子レベルでの相互作用は、突き詰めると水素結合に由来する。タンパク質の場合、アミノ酸の側鎖が整合して水素結合を形成する。もちろん、いくつも水素結合があればより強固な相互作用となる。細胞の中では、一時的(トランジエント)な相互作用や常にくっついているものまで様々だ。さらに、翻訳後修飾(PTM;Post Translational Modification)にも注意が必要で、シグナル経路に代表される相互作用にはリン酸化された(あるいはされてない)時のみ相互作用ができるものもある。それらを全てマップするのは膨大な組み合わせになるため、依然として多くの研究がなされている。

そのような相互作用を実験的に観測することは手法の時間解像度などの問題から中々難しい。近年の計算機(スーパーコンピュータ)の進歩で、原子レベルでのシミュレーションが可能になりつつある。実際の分子がどのように挙動しているかをMDシミュレーション(Molecular dynamics simulation)の動画で見てみよう。
1. タンパク質間相互作用データベースにアクセスしよう
タンパク質間相互作用データベースのBioGRIDにアクセスしよう。
BioGRIDは様々な文献で報告されているタンパク質間相互作用を統合しているデータベースで、オンライン上で任意のタンパク質について報告されている相互作用を確認することができる。
ここで注意が必要な点をいくつか挙げる。
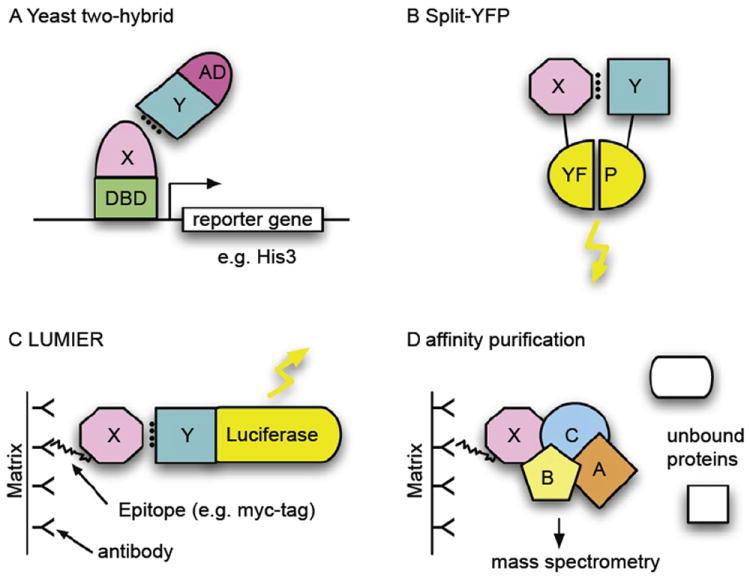
- タンパク質間相互作用は様々な手法を用いて同定されるため、同定された手法に気をつけよう。
最も確実かつ情報量が多いのはCo-crystalizationなどが挙げられ、次にBinaryの相互作用を検出できるY2HやPCAがある。免疫沈降ー質量分析(AP-MS)法はタンパク質を芋づる式につってしまうので、精査が必要だ。
- タンパク質ペアの数は膨大(ヒトでは 20,000 x 20,000 / 2 = 20,000,000 ペア)で、全てが試験されているわけではない。あくまで、「これまでに報告のある」相互作用しか載っていない。
システマティックに全ての相互作用をスクリーンする仕事でない場合は、特定の遺伝子やタンパク質に着目したアッセイが行われる。そこでp53などの癌促進遺伝子などが他と比べ多く報告されることで、実際の相互作用数を表さないような分布を示すことがあるので気をつけよう。
2. 着目するタンパク質について、その相互作用を確認しよう
BioGRIDのページにアクセスし、自分の着目するタンパク質をサーチしよう。種がわかっている場合はプルダウンで種を指定しよう。ヒットしたタンパク質の一覧が出てくるので、自分の着目したタンパク質と合致したものを選択する。
Interactorsのタブから、相互作用の報告されているタンパク質一覧が表示される。右のDetailsをクリックすると、報告の内容(論文、手法等)が見られる。今回の演習では立体構造の解かれているものを選びたいので、Co-crystal structureの報告があるかを見てみよう。
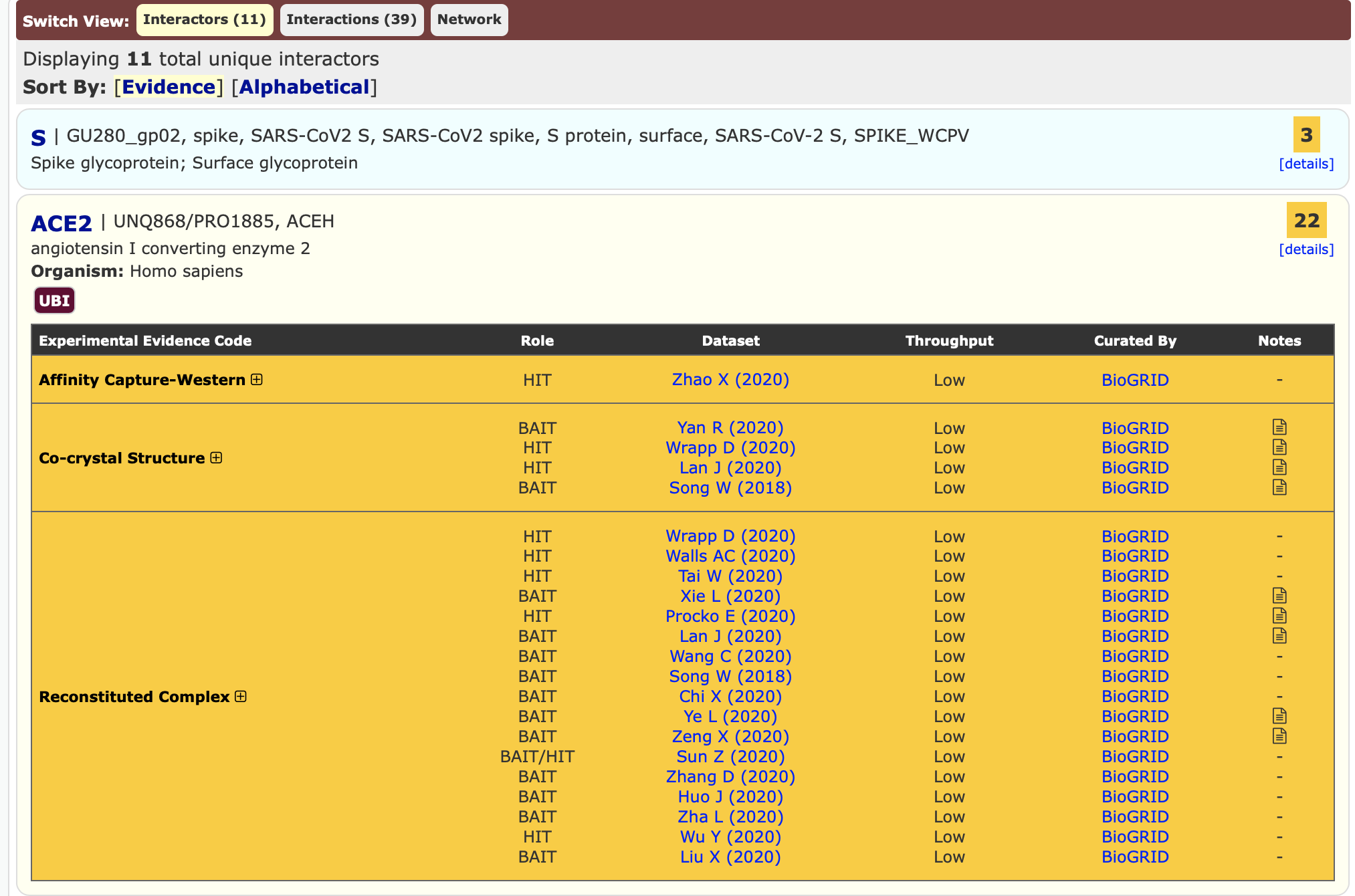
SARS-CoV2では、Spike protein (S)がヒトのACE2と相互作用することが報告されていることが確認できた。
3. 立体構造の登録のある相互作用を見てみよう
ここで第2回の演習を思い出し、PDBで検索しよう。大抵は「PDB 着目したタンパク質 相互作用していたタンパク質」とググればトップにヒットする。相互作用をしているタンパク質について、3D veiwで表示しよう。ここで、第2回で確認したバリアントの位置を照らしてみよう。
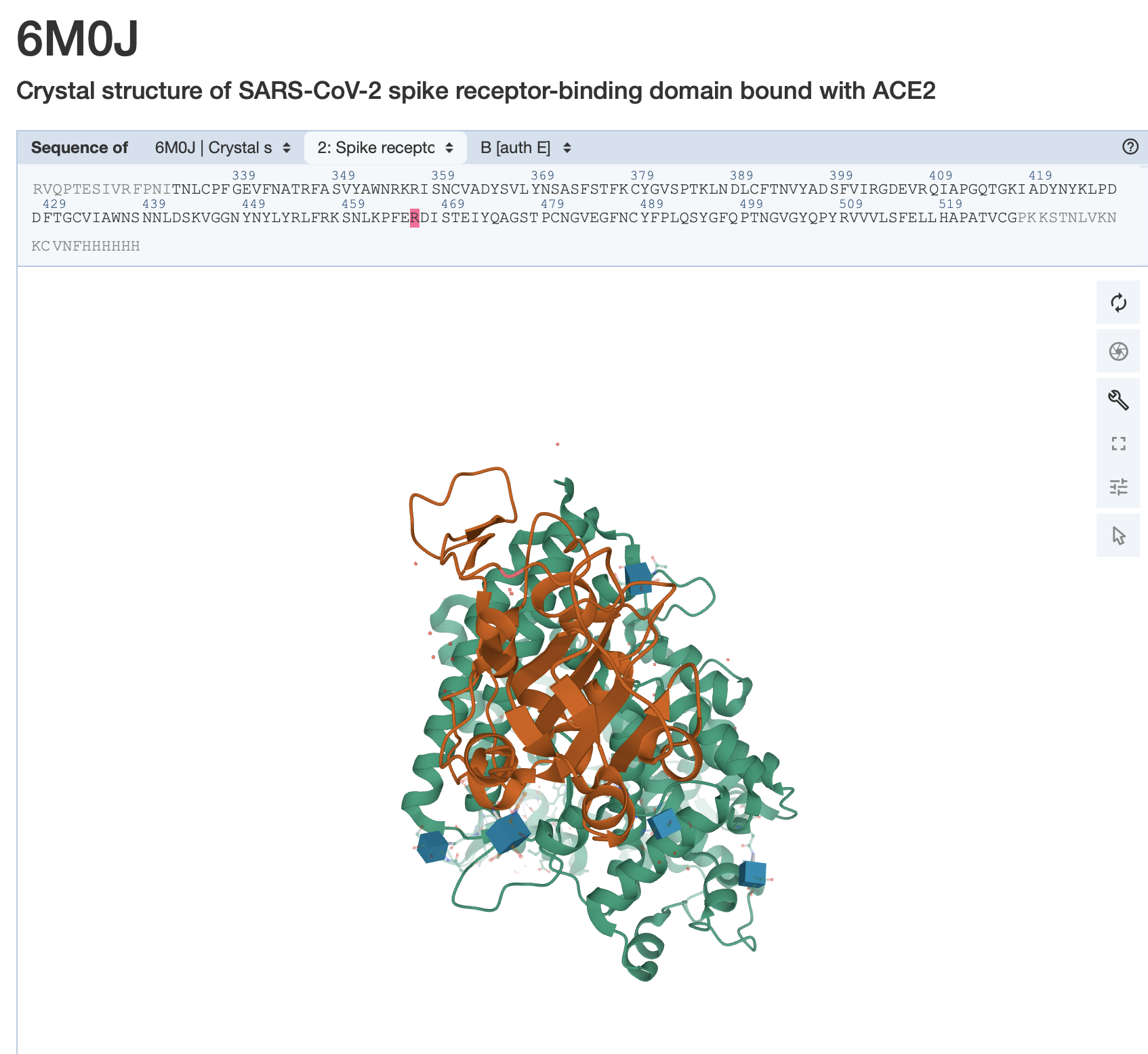
この結果のスクリーンショットをSFC-SFSで提出しよう。また、良く保存されている領域はどのようなところだろう?
まとめ・Take home messege(全3回)
今期の生命動態のデータサイエンス山本担当の3回では、タンパク質の相互作用と変異情報をもとにバリアントの機能的差異について考察した。その過程で、各種データベース(ゲノム、タンパク質立体構造、タンパク質間相互作用)のから自分の必要なデータの取得、グローバルアラインメントを演習形式で学んだ。本演習では、それぞれ各一つのタンパク質に着目したが、例えば相互作用相手の変異やホモログ等を考慮するとさらに多くの解釈ができてくるだろう。今後、配列解析等をする際はその配列の差異が生物学的にどのような違いに紐づくかについて頭を巡らせてほしい。
短い間でしたが、拙い講義にお付き合い頂きありがとうございました。よろしければ講義アンケートから率直な感想を教えてください。今後の講義などに役立てさせていただきたいと思います。